1. Introduction
In the previous chapter we discussed the Particle-Mesh method. This method is
relatively simple with respect to software engineering, but mathematically a
little more complicated (at least in my perspective !).
With the Tree-Code method we have in fact quite the opposite problem : from
a mathematical point of view this is quite a straightforward method, so there
won't be many nasty formulas in this part, but the necessary dynamic memory
management and recursion makes software development not completely trivial.
Tree-code methods are a completely different way to achieve the same result
as with Particle-Mesh : downscale the computation time for a simulation to be
of order N·logN instead of N2 .
The basic idea is to replace a faraway group of particles by only one
virtual particle, exactly located in the centre of mass of this group and with
a mass that equals the reduced mass of this group of particles.
This virtual particle is a particle that obviously attracts with the same force,
no matter in which direction we are looking. Such a center of force is often
called a monopole. The first implementation was published about 11
years ago in Nature in an article by Joseph Barnes and Piet Hut (see []).
Under some circumstances (for example with a group of particles that is very
long in one direction but short in the other direction) the approximation with
only one particle could not be good enough. In this case a so called multipole
would be used, which sounds heavy but in fact is nothing else than using 2,
3, 4 or even more virtual particles instead of only one. (for the example in
this paragraph a dipole would be a good approximation).
The extension from Monopole to Multipole is known as ``Fast Multipole
Method'' and it's basics were first published by L. Greengard and V. Rokhlin
about 10 years ago( see []).
Main difference is that the FMM method doesn't store the mass but the potential
for a certain cell and as such it may be viewed as a sort of ``cross-breading''
between PM and pure Barnes-Hut (BH). Because of this potential it is possible
to use FFT techniques just as with PM and this can make the FMM even faster
than BH. Most people state that the FMM can scale with order N .
As the BH algorithm is relatively simple to understand and because FMM may be
viewed as just one of the many possible optimisations of BH, I choose to forget
about FMM for the time being and just investigate and discuss BH. If you understand
Barnes-Hut and saw Particle-Mesh it is not so difficult anymore to get the essence
of FMM, while understanding FMM from scratch is a very hard thing to do.
So let's see in a little more detail how Barnes-Hut works and what kinds of
practical problems I ran into when trying to implement it using the article
from Nature ([]) as a starting point.
2. Algorithm based on Barnes-Hut method
|
- calculate the total energy and momentum
|
|
- initialise the tree (rootcell dimensions, initial memory)
|
|
- while ( timecounter < endtime )
|
|
- sort all particles on either the X or Y coordinates
|
|
- recursively split the roottree into 4 daughter cells until every
|
|
particle fits into a separate cell (top
® bottom)
|
|
- recursively compute the total mass and centre of mass for
|
|
all cells in the tree (bottom ® top)
|
|
- count all particles in the rootcell (using the sortlist) to be
|
|
sure no particle moved outside the tree
|
|
- for all particles do
|
|
- recursively compute force contribution of all cells starting
|
|
at the rootcell using the following rules :
|
|
- count the number of particles ( n) in the current cell
|
|
- if n = 0 do nothing (empty cell)
|
|
- if n = 1 compute force contribution of this single particle
|
|
(unless it is the currently processed particle !)
|
|
- if n > 1
|
|
- compute the distance r to
the centre of mass and the cellarea A
|
|
- if ( r/A > q ): cell is faraway so compute force contribution
|
|
using total mass and centre of mass and go to next particle
|
|
- else: recursively process the subcells down the tree until
|
|
1 particle in cell or far enough away
|
|
- endif
|
|
- end recursion
|
|
- calculate new speed and position using a suitable scheme
|
|
- endfor
|
|
- write or plot the new particle positions
|
|
- increase timecounter with the timestep
|
|
- endwhile
|
|
- check the energy and momentum for conservation
|
Table 1: Flowchart Tree-Code for a 2D system
- As said before : the essence of the Barnes-Hut algorithm is to discriminate
between faraway and nearby particles and to do the timeconsuming Particle-Particle
computation only for nearby particles. To achieve that we first choose an initial
areasize, large enough too ensure no particle will leave the area during the
whole simulation. This initial area is our ``rootcell''.
- Now we start splitting up this rootcell into 2dim daughtercells ( dim : dimension:
either 1, 2 or 3D, resulting in either a binary-, a quad- or an octtree) until
we have captured every particle into a separate cell. Obviously for dense areas
it will take a lot of splitting up until everything is separated, while in sparse
areas there almost won't be any splitting at all : the tree adapts itself to
the local complexity !
Sometimes this is called an adaptive grid, although it is important to realise
that here we do not use a spatial discretisation, so the term grid
is a little misleading in this case. A tree example for the 4-body problem from chapter
is shown in figure .
Im my implementation I had to count the number of particles in each daughtercell
after every split to find out if it was necessary to split that cell any further.
Initially this took a lot of the total computation time, but after some experiments
with sorting techniques I was able to decrease the time spent for counting considerably
(see ).
Later I found out that most of the ``guru's'' use another technique : insertion
of the particles in the tree and split up the cell as soon as it already contained
at least one other particle (in fact I found no one who used my sorting method
.... ), but as my implementation worked reasonably well after adding the sorting
optimisations I decided to be stubborn and to continue using my own method,
following my favourite maxim that : ``it is better to have your own
algorithm than a good algorithm !''
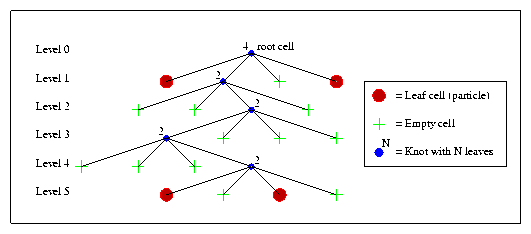
Figure 1: Tree example for the 4 body problem from chapter 1.3.
- Note that building the tree was started at the top and ended at the bottom.
Now it is time to compute for each cell the reduced mass and the centre
of mass, and this is much better done starting at the bottom and ending at
the top: doing that we could use the information of the lower level in the tree
to compute the information for the level just above that.
This trick ensures that we only have to compute the contribution of at most
2dim (virtual) particles as that is the maximum number of daughtercells each cell
can have : in for example a 2D problem the centre of mass of a parent cell can
be computed by only taking the (centre of) mass of it's 4 daughtercells,
without knowing anything about the level below the daughters (or sons if you
like) !
At the lowest level of the tree (in the so-called leaf cells) things are even
more simple: there we only have to use the mass and position of the single particle
in it and thus it is not necessary to compute or even assign reduced
mass and the centre of mass in this case: just a reference to the
particle information will be sufficient.
- Now the tree is completely finished. But as we have to do the steps above for
each iteration it may be a good idea to count all particles in the root cell,
to be sure we really have all particles included. This is because it is always
possible that a particle moves outside the rootcell dimensions, which could
have nasty results for the simulation !
-
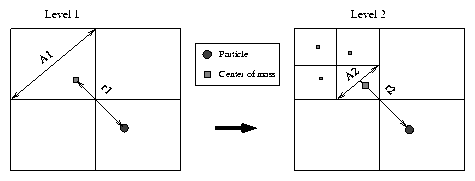
[r1/A1] < Q: too close Þ go one level deeper [r2/A2] > Q : this is fine
Figure 2: Discrimination between faraway and nearby cells using Q = 1\protect (note that r2 » r1\protect )
If the tree is ready we can turn to the most important part : determine how
large the total force is that works on each particle. This is again
done in a top-bottom approach: for every particle we start at the rootcell and
then go down the treebranch until we have reached the lowest point. Every cell
we pass contains either 0, 1 or more than 1 particles, each of which asks for
a different treatment :
- n = 0 : The cell didn't contain any particles and can just be ignored.
- n = 1 : The cell contains one particle : in that case we can just calculate the
exact contribution of that single particle to the total force on the particle
currently being examined, just as in PP.
- n > 1 : The cell contains more than one particle. Now comes the most difficult part,
but also the part that can make TC so much faster than PP : we have to find
out if this cell is far enough away to approximate the force by the reduced
mass and centre of mass of this cell: the cell is being used as a virtual particle.
This was very cleverly devised by Barnes and Hut : they introduced a special
parameter Q, that should be compared with Qp = [r/A] , where r is the distance from the
particle being examined to the centre of mass of the cell
currently being examined (not the cell-centre !) and A is a measure
for the size of the cell at the current level. The exact value for Q is the
discriminating factor that decides who is far enough away and who is too close.
If the cells are all square or cubic as in BH, then A seems to be equal to the
size of only one edge, although unfortunately the article by Barnes-Hut
isn't very clear at this point. But because I used tree cells that could have
different dimensions in each direction (which can be dangerous, but
also gives the user some extra freedom in case of special problems) I decided
to replace this to use the diagonal A = Ö{w2+h2} instead.
This yields us Qp2 = [(r2)/(w2+h2)] , where w and h are the cell dimensions.
Note that this results in my value for Qp for a square (or cubic) cell being
Ö2 (or [Ödim] to be accurate) smaller than in the case of BH.
There are obviously 2 possibilities :
- The first case in 1 simply leads to the conclusion that the cell is far enough
away to use it as a virtual particle without looking any further at its daughtercells.
- The second case in 1 shows that the cell is too close and that we should go
down one level and repeat everything starting with point 2 for each
of the 2,4 or 8 daughtercells. How exactly the factor Qp changes as we go to
a lower level and thus how Q (which is constant during the simulation !) can
separate faraway and nearby cells is shown graphically in figure 2.
- If we have calculated the total force on all particles, we can use again one
of the integration schemes already discussed in chapter
and calculate new positions and speed.
- And then we can increase the timestep and start again at 2 where we have to
repeat everything again, including building the tree and computing the reduced
masses and centre of mass.
The whole algorithm is described in the flowchart in figure 1.
3. Choice of the Q\protect parameter
As should be clear from above, the choice of the parameter Q is very important.
The larger it will be, the further away the cell has to be, before we can use
the virtual particle approach and thus the more the algorithm will behave like
PP: accurate but slow. For Q® ¥ TC simply turns into a very complicated way to
do plain old PP.
But how small can we make Q before running into real problems ? That is not
really difficult : we should avoid under all circumstances that we use a cell
from the same branch as the particle we want to know the total force on, as
in that case the mass of the examined particle itself can contribute to it's
own acceleration ! (usually called self-acting forces) So, basically all
cells from the branch where the particle belongs to are forbidden, no
matter how far away or how large they are, except of course for the lowest
level, where only real other particles are found.
In theory, with the examined particle having a mass of almost 0 and with very
heavy other particles at a strategic point in the same cell, there is small
chance that the centre of mass for the cell lies in the upperleft corner, while
the examined particle is in the lowerright corner. In that case the distance
will be r » Ö{w2+h2} = A and thus Qp will be approximately 1 . (although still slightly smaller
than 1 !) So, as long we keep Q > 1 , we can be sure to avoid the self-acting forces
under all circumstances, at least with my definition for Qp .
Note that in the case of the definition for Qp as given by Barnes and Hut Q should
be bigger than Ö2 to be on the safe side.
One more remark: unfortunately I choose the definition of Qp to be just the opposite
as in the Barnes-Hut article (stating Qp = [A/r] ), so where BH says Qp < Q I say Qp > Q and vice-versa.
Sorry for the extra confusion this may cause, but I only found out this when
I was almost finished with the report and changing the code at this point doesn't
seem a good idea to me.
Resuming :
- Values for Q < 1 are strictly forbidden as they can cause self-acting forces (but
this happens only in rare cases).
- Large values for Q yield a more accurate simulation, but also causes a longer
computation time.
I choose the Q to be default 1. To give an idea about the effect of variations
in Q, I ran my TC program on a random set of 500 bodies , using 50 iterations
and leapfrog (Linux on a Pentium 75) :
|
Discriminating factor Q2 |
1 |
5 |
10
|
|
|
|
Energy leak over 50 iterations (%) |
-0.336531 |
-0.336423 |
-0.336426
|
|
Runtime for 50 iterations (sec) |
15.51 |
34.92 |
49.24
|
|
Momentum leak X direction (%) |
-0.77 |
-0.03 |
0.001
|
|
Momentum leak Y direction (%) |
-0.248 |
-0.008 |
0.005 |
As was to be expected the runtime increases rapidly with increasing Q but as
a compensation the accuracy improves at the same time.
In normal situations a value of 1 gives acceptable results with reasonable speed.
Recently it was however shown by Salmon and Warren ([]) that it is
possible for the error to become unbounded in some cases, so it is always wise
to pay a little attention to this point. They also give some other possibilities
for the definition of Q that can be interesting, but that are not further discussed
here.
4. Implementation aspects
4.1 Sorting techniques
As said before: my implementation for TC is slightly different from most others
as I found out later. This is caused by the fact that by reading the article
you only get the basic idea of an algorithm, so the practical problems is something
you have to solve by yourself and sometimes you (or in this case I !) simply
do not make the best choice. Let's view this difference a little closer :
- Barnes-Hut just loops through all particles and inserts each particle in the
daughtercell that is currently on the lowest point in the appropriate branch,
splitting up that daughtercell as soon as it contained more than 1 particle
after the insertion. This has the advantage that it is not necessary to count
how many particles there are in a cell, as when the tree is finished and a counter
is updated every time a new particle is inserted the total number would be automatically
correct.
There is however some extra work to be done as we need to determine in what
daughtercell (that should be always at the bottom of a branch) the particle
should be inserted. This makes it necessary to walk through one complete branch
of the tree for each particle.
- My Tree-Code version starts at the root cell, counts all particles in the cell
and splits recursively into a number of daughtercells, where again all particles
in each daughtercell have to be counted. This counting involves comparing the
coordinates of each particle with the current daughtercell dimensions.
This counting determines whether or not we have to split up the cell any further
and as such is a crucial step in the program.
How important counting really was I only discovered after some analysis with
the fantastic profiler option provided by the GNU-C compiler (-p) : I found
that most of the total computation time was spent on comparing the coordinates
if I ran a sufficiently large simulation ! As this made my program incredibly
slow I started thinking of something to speed things up and decided to use a
sorting algorithm for these coordinates.
I choose to sort only on either X or the Y-coordinate, as this could already
reduce the number of particles to be examined considerably. If you know that
the particles are sorted on one of their coordinates (how this could be done
will be discussed later), the following procedure could be used to compare the
coordinates with the cell dimensions (let's assume the particles are sorted
on the X-coordinate) :
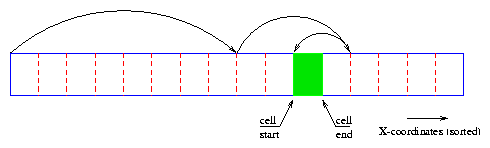
- Search the first particle in the sortlist that has an X-coordinate
larger or equal than the X coordinate of the left edge of the cell ( xn ³ xl ).
- From here you can start comparing the Y-coordinate of each particle
and check if it is larger or equal than the lower edge and smaller than the
upper edge and increase the particle counter for each particle that fits this
condition ( yn ³ ybÙyn < ytÙxn < xr ).
- You can repeat the previous step until you reached a particle with X-coordinate
larger or equal than the other edge of the cell ( xn ³ xr ).
- As soon you find such a particle you can be sure that all remaining particles
in the sortlist are outside the cell and so you could stop counting the particles
for this cell.
Note that the differences between ``larger than'' and ``larger or equal'' are very
important, as this is the only way to avoid particles being included in 2 cells
if they happen to be exactly on the border between them.
In an average distribution you can assume that the number of particles to be
compared in step 2 is about the square root of the total number so this method
indeed can be a time saver.
But a few problems are still left to be solved :
I think the difference between Barnes-Hut and my method - with the improvements
described above - wouldn't be very significant, but I have to admit I didn't
check this yet and so it remains to be done. So there is still a possibility
that my method is faster .....
4.2 Memory considerations
- Empty cells
- As could be seen in picture 1, highly non-uniform problems could
result in a considerable amount of empty daughtercells (leaves). In some descriptions
of Tree-Code these empty cells are removed from the list to avoid them wasting
memory.
I made removing empty cells a compile-time option and to my surprise I found
that removing the empty cells makes the algorithm in some cases faster instead
of slower !
This could be explained by the fact that - although removing empty cells takes
more work in building the tree - the possibility to skip them immediately in
the Centre-of-Mass computation and in the computation of the acceleration can
save quite some time. The latter only happens of course if there are enough
empty cells to be removed from the list or else we'd just do extra work for
nothing !
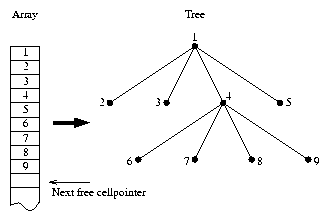
Figure 4: Cell array to cell tree mapping.
- Dynamic tree allocation
- If we would do a malloc() for each new cell
and do a free() for the whole tree after every iteration, most of the
computation time would be spent in memory management as I discovered. On the
other hand : using an array of static dimensions would lead to problems as it
is difficult to predict how many cells would be necessary to build a tree for
a certain problem (see figure 1 for a bad example !), so this is also not a
satisfactory option.
I decided to use a ``best of both worlds'' approach: I malloc an initial
array of cells of length N and as soon as I run out of cells at a certain point
in the program, I do a realloc to increase the array with again N elements.
This ensures the array will allways be large enough to supply all the necessary
cells for a certain tree.
A decreasing/increasing ``nextfree cell'' counter is used to decide which cell
is going to be the next one to be used to build the tree.
Note that storing pointers for the daughtercells in the tree is not
a good idea : after each realloc the whole array may be (but not has
to be !) shifted to another memory location, making the pointers useless. The
only good way is storing the relative offset to the start of the array (the
rootcell). This took me several hours to find out, so a warning at this point
doesn't seem to me a complete waste of paper !
The relation between memory and tree is shown in figure 4.
- Cellsize
- The dimensions of each cell can either be stored in the cell record
or be computed every time it is needed by using it's level in the tree. The
first option uses more memory, the second takes more computation time. I have
put the choice in a compile time parameter, so both can be used.
4.3 Tunable parameters
As with PP and PM, there are many variables that can be used to alter the specific
behaviour of the simulation. In the datafile the stepsize, the number of iterations
and the number of iterations between a screen update can be specified. For the
exact meaning of the datafile layout, see the appropriate appendix in .
Commandline options :
00.00.0000
- [
-
-i,a,x,f,o]Same function as with PP (integration method, animation, no graphics,
no energy error computation and output coordinates to a file).
- [
-
-w < nr > ]Width of the root cell, expressed in the
same units as the particle positions.
- [
-
-h < nr > ]Height of the root cell, expressed in the
same units as the particle positions.
- [
-
-t < nr > ]Value for the discriminator Q2 (default equals
to 1).
- [
-
-p]Plots the new tree after every screen update. Gives a nice idea how the
tree is built , especially if combined with the animate (-a) option.
- [
-
-u < nr > ]It is not absolutely necessary to update
the tree after every iteration. With this parameter the number of iterations
between 2 tree updates can be changed. It is set to 1 by default. To give an
idea of the influence of this parameter I ran it twice with u=1,3 and 10 on
the now wellknown set with 4 particles and a random set of 500 particles (Linux
on a Pentium 75) :
|
4 particles: |
u=1 |
u=3 |
u=10
|
|
|
|
Runtime (7580 iterations) |
3.02 |
2.16 |
1.66
|
|
Energy leak (%) |
0.061 |
0.063 |
0.067
|
|
Momentum leak (X) (kg.m/s) |
-10.96 |
-224 |
-957
|
|
Momentum leak (Y) (kg.m/s) |
-15 |
-57 |
-124 |
|
500 particles: |
u=1 |
u=3 |
u=10
|
|
|
|
Runtime (50 iterations) (%) |
15.35 |
14.25 |
13.48
|
|
Energy leak (%) |
-0.3365 |
-0.3364 |
0.06145
|
|
Momentum leak (X) (kg.m/s) |
42.06 |
47.67 |
-10.96
|
|
Momentum leak (Y) (kg.m/s) |
-12.2 |
-12.6 |
-15.41 |
00.00.0000
- [
-
-d < file > ]Write the initial tree to a file and exit.
Only useful if combined with -o option: The two files can be used to build a
picture of the initial situation at the start of the simulation. (i.e. to use
it in this report :-)
At compile time there are several other parameters that can be set :
- NSUB
- This sets the number of daughtercells for each parentcell. A value of
2 yields a binary tree, 4 a quadtree and 8 an octtree.
- BLOCKSIZE
- This is the initial size of the tree array. Every time the system
runs out of cells, the array will be increased with BLOCKSIZE new cells. If
it is set very low, the memory allocations may take up a significant time at
the first iteration. However, as the array is only enlarged and never shrinked,
this will only affect the first iteration.
Note that if two particles come very close the array size may increase very
fast: it may take quite some splitting before the two are separated.
- NO_EMPTYLEAVES
- This removes all daughtercells that contain no particles. Removing
them takes more time, but then these leaves can be skipped in the centre of
mass computation and the force evaluation. This is the default as in most cases
it is the fastest method.
- RECALC_CELLSIZE
- The exact cell dimensions can be directly computed from the
size of the rootcell and the level the cell belongs to: in 2D a cell at level
2 has exactly [1/4] the size of the cell at level 1. Computing the cellsize each
time it is needed instead of storing it for each cell saves memory, but increases
the computation time.
5. Collision management
Of course the collision management from PP works fine here, but there are 2
remarks to be made :
- In theory it is possible that a collision takes place between a real particle
and a cell not being a leave (a virtual particle). Although this can
only happen if Q is set to a much too small value, I only report the fact that
a collision has occurred, but not between which particles to avoid confusion.
- Non-elastic collisions by maintaining the 2 collided particles at the same place
may cause large troubles in building the tree : it is impossible now to separate
the 2 particles into different tree-cells, no matter how often we subdivide
the tree !
6. Visualisation techniques
The same graphical interface as used for PP is also used for the TC program,
so the remarks in the PP chapter also apply here. The only addition is that
also the tree itself is plotted and even can be animated if necessary, which
yields quite a spectacular picture, at least in my opinion !
(use the command : treecode -a -p <parfile> to get this animated tree)
7. Restrictions
For PM the restrictions were fairly straightforward : only uniform distributions
with a large number of particles. For PP things were comparably straightforward
: use it only for a limited number of particles or where accuracy is of major
importance.
For TC it is slightly more complicated : TC produces most of the time very accurate
solutions in a reasonable time. But unfortunately this is not always the case,
as shown in []. It only is a bit difficult to find out if the worst
case situation as described in their article may arise while investigating a
certain problem. Methods like I used in my analyser (auto-correlation) (see
) are not able to say anything useful about this.
An indication may be the error in the energy and the momentum, but even there
it is possible that cumulative errors may cancel each other partially, especially
in the energy where only RMS (Root Mean Square) or average errors occur.
On the other hand, the solutions suggested by Salmon and Warren may increase
the computation time so dramatically, that it sometimes simply doesn't seem
worth the trouble in most cases.
So I think we can say that TC produces reasonable results as long as we bear
in mind that these worst cases - although rare - may occur from time to time.
If we really want to be on the safe side Salmon and Warren suggest to use a
small Q : less than 0.5. Because of the differences between my definition of
Q and the one used by Barnes-Hut and Salmon-Warren this implies for my method
Q > [1/(0.5·Ö2)] = Ö2 or Q2 > 2 .
An indication of what this means for the computation time can be found in the
table from section 3.
File translated from TEX by TTH, version 1.23.