Chapter 6
Analysis: Which method for what problem ?
1. Introduction
In the previous chapters we have discussed in detail the properties of 3 basic
particle simulation methods and we have seen what their typical advantages and
disadvantages are. But now comes the question what method should be chosen for
a certain problem to obtain an optimal result.
In the literature about particle simulations there was surprisingly little I
could find about this topic. There is a lot being said about collisionless and
collisional systems, but how to distinguish between these two types given a
certain dataset is a question that is left largely unanswered. But exactly this
is the most crucial point in deciding which method would be best suited. I started
thinking about this subject myself and tried to do the following :
- Determine which parameters/properties of a given dataset could be used to make
a reasonable decision.
- Design a simple algorithm - or better : a heuristic - that could be used to
make the choice without the need of many complicated computations. Using all
3 methods and see which one gives the best results is certainly not the best
option ...
- Write the appropriate code, that should be fast enough to be able to give a
clear indication for even very large datasets within reasonable time.
2. Parameters and properties
If we need to distinguish between parametersets so to be able to make the right
choice, what parameters could then be used to make the best decision ? When
I tried to tackle this problem I started to think of several characteristics
that might be interesting enough to be used to predict something about the simulation.
I'll discuss the most important ones briefly hereafter :
- The number of particles ( N ). It may sound a little trivial, but of
course if the number of particles is lower than a certain threshold it is useless
to use either a Tree-code or a Particle-Mesh method, as Particle-Particle is
definitely the most accurate and in that case undoubtedly also the fastest choice.
- The distances between each particle and it's closest neighbour ( MD )
and the variation in these distances. For each particle we can compute the distance
to all other particles. The shortest one found is the minimum interparticle
distance ( MD ) for that single particle. The shortest value for all these
MD values is the shortest minimum interparticle distance ( SMI ) and the
average of all these values is the average minimum interparticle distance
( AMI ).
The difference between the shortest minimum interparticle distance
( SMI ) and the average minimum interparticle distance ( AMI ) can give a rough
indication for the uniformity of the mass-distribution. Note however that 2
very close particles in an otherwise reasonably uniform distribution can give
very unreliable information about the uniformity of the particle distribution.
Also note that this value doesn't say anything about the density distribution,
unless all particles have the same weight !
- The method mentioned before can be improved by computing the standard
deviation in the minimum distances ( MD ) found for all particles. A large standard
deviation implies a large variation in these minimum interparticle distances,
while a small one implies a more or less uniform distribution. Note that for
a grid of particles - which could be considered as very uniform - the standard
deviation should be always 0 !
- The potential and kinetic energy and the ratio between them. The potential
energy is a measure for the amount of acceleration that would occur during a
simulation. The kinetic energy on the other hand says something about the velocity
of the particles. If the kinetic energy dominates, the approximated displacement
during the whole simulation can be computed using only the kinetic
energy and the intended duration of the simulation.
If on the other hand the potential energy dominates we could give an indication
about the displacement by only using the initial acceleration and assuming
it would be constant during the whole simulation.
- The mass (density) distribution and it's autocorrelation ( DA ). The density
distribution can be obtained in a similar way as with the Particle-Mesh method.
If we use this result to compute the autocorrelation function of the
density distribution (see hereafter) we can obtain information about the mass
uniformity.
If the system appears to be more or less uniform according to this observation,
the Particle-Mesh method could be used, provided the ratio between the average
minimum interparticle distance and the average displacement ( AMI/AD ) is large enough
to guarantee that many collisions are unlikely (see hereafter).
- The average displacement ( AD ) and it's variation during the intended
time of the simulation. If we have an indication of the size of the per-particle
displacement, we know also how likely collisions can be if we combine it with
the average minimum inter-particle distance from above.
- The ratio between the average minimum interparticle distance ( AMI ) and
the average displacement ( AD ). The combination between the average minimum distance
and the average displacement can give useful information about the number of
collisions that may occur during the simulation.
This can make the distinction between collisional and collisionless systems
and thus between the choice for either Tree-Code or Particle-Mesh, but of course
this only makes sense if the variation in either the average minimum distance
and the average displacement is not too large.
In a distribution that was already found to be reasonably uniform, this could
be useful extra information to improve the quality of the decision.
- Number of detected collisions during one simulation step. What we actually
do is compute one timestep using a Tree-code like algorithm. In this simulation
collisions can occur. If we find a significant number of collisions within this
single timestep Particle-Mesh is certainly not the best option. Note that it
may also be wise to reconsider the timestep in this case ....
Many of the observations mentioned above were combined into a piece of software
that is able to give an indication about the properties of a certain dataset
and it also can give a hint what method would be best suited according to these
observations. The complete heuristic as was implemented in my analyser program
is shown in figure .
Picture Omitted
Figure 1: Flowchart for analyser program.
3. Autocorrelation
A very important part of the program is the computation of the autocorrelation
function. It can give us valuable information about the uniformity of the interparticle
distances or about the density distribution and that is why I want to give it
a little more attention here.
The autocorrelation of a function f(x) can say something about how regular this
function f(x) is over a certain interval Dx : it is the amount of ``overlap'' we find
if we shift the function over a distance Dx to the left or the right. We just
measure how large the overlapping area is for a certain value to find the autocorrelation
for that distance.
A large overlap indicates a large auto-correlation for this value of Dx and a
small overlap on the other hand means a small auto-correlation for Dx .
Of course there exists for every value of Dx a corresponding value for the ``overlap''
and all these values together gives us a new function : ``the autocorrelation
function'' A(f(x)) for our function f(x) .
An autocorrelation function can be computed in the following way (note that
I write functions of 2 variables now as that is what we are interested in for
our analysis) :
|
f(x,y) Þ A(f(x,y)) = |
ó
õ
|
l
|
|
ó
õ
|
k
|
f(x,y)·f(x+k,y+l) dk dl |
| (1) |
If we define a helper function g(x,y) = f(-x,-y) we can rewrite this equation into a (2-dimensional)
convolution of the functions f(x,y) and g(x,y) , which is hopefully known to the reader.
(if not you may find more about convolutions in [] or [])
This yields us for 1:
|
A(f(x,y)) = |
ó
õ
|
l
|
|
ó
õ
|
k
|
f(x,y)·g(k-x,l-y) dk dl |
| (2) |
Agreed, this is still not really pleasant to look at, but this will change if
we transform the equation above into the (2D) Fourier domain, just as we did
with the Poisson equation in chapter :
|
|
~
A
|
(f(x,y)) = |
~
A
|
(k,l) = |
~
f
|
(k,l)· |
~
g
|
(k,l) |
| (3) |
Equation 2 then changes into a straightforward multiplication of the two (transformed)
functions [f\tilde] and [g\tilde] .
3.1 Discretisation
As with the Poisson equations before, the functions f and g could be replaced
with a set of numbers : the values of f and g at the gridpoints that specify
the density distribution. The method to setup the density distribution is exactly
the same as with the Particle-Mesh method.
That gives us the possibility to use the FFT again to obtain the autocorrelation
distribution: transform the distributions f and g into the distributions [f\tilde] and
[g\tilde] , then multiply all corresponding values of [f\tilde] and [g\tilde] with each other and finally
go back to the spatial domain again using an inverse FFT.
Special attention needs to be paid to the spacing and the dimension
of the grid :
The gridspacing should be of approximately the same order as the mean
interparticle distance or else we either wash out information or introduce high
fluctuations in the autocorrelation function. I choose to make it about twice
the size of the average minimum interparticle distance to smoothen the function
somewhat and also to reduce the computation time. The gridspacing is also limited
by the number of particles: it is useless to have a number of gridcells that
is several magnitudes higher than the total number of particles.
As for the dimension of the grid : to avoid aliasing it is important
that the grid is more than large enough to contain all particles. I calculate
the maximum interparticle distance in both directions and set the grid size
at about 4 times the size of the particle space.
4. Uniform and non-uniform problems
To test the software packages, I used a number of datasets, some of them designed
earlier for the Parallel Computation Practice. However, for the larger problems
( > 103) I didn't want to keep the datasets as they tend to occupy a lot of diskspace.
For this reason I built 2 small helper programs:
- mkgrid :
- A dataset generator for monotonous grid patterns.
- mkrnd :
- A dataset generator for random patterns.
Both helper programs can also generate random initial speeds if desired. For
the random sets applies that the seed for the random generator is always 1,
so this should result in every time the same pseudo random sequence if called
with the same arguments (if everything works well).
Let's take a closer look now at the differences and similarities for the patterns
being generated by these helper programs.
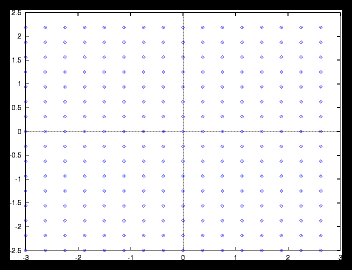
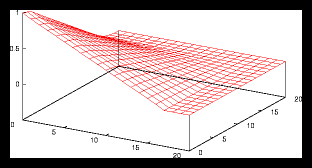
Figure 2: A small gridpattern and it's autocorrelation function (256 particles)
For the gridpatterns we can say that they are always uniform, no matter how
large the problem is, provided of course that the masses are identical. To see
what this means for the autocorrelation function of these patterns, I have plotted
a relatively small one and a larger one, together with their autocorrelation
functions (see figures 2 and ). Apart from a few scale factors you can see
that they are virtually the same.
Note that it is not a good idea to have a smaller spacing on the autocorrelation
grid than the meshspacing itself, or else we'll see large fluctuations for the
autocorrelation function: some cells will be empty while others will still contain
1 particle.
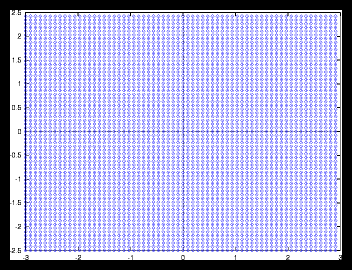
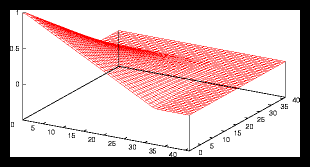
Figure 3: A larger gridpattern and it's autocorrelation function (4096 particles)
For the random patterns (datasets where the positions are generated at random),
we'll see that the results will be a little different. Examples can be found
in figure and . Now the smoothness of the autocorrelation function also depends
on the gridsize that is used to determine the density distribution !
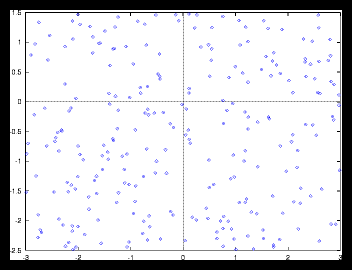
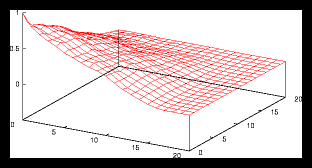
Figure 4: A small random generated pattern and it's autocorrelation function (256 particles)
This is caused by the following effect: as soon as the size of a gridcell is
large compared with the average interparticle distance, we'll find approximately
the same amount of particles in each cell, no matter at which cell we are looking.
But if the size of a gridcell is too small, we'll see large variations as the
number of particles per cell is going to vary much more in that case than with
a monotone grid.
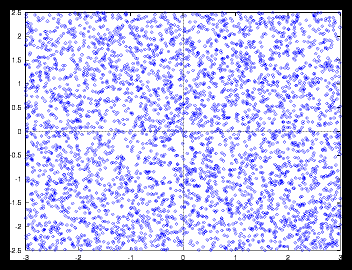
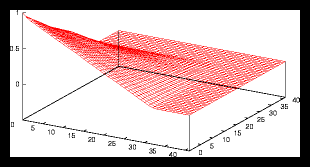
Figure 5: A larger random generated pattern and it's autocorrelation function (4096
particles)
Nevertheless we can see that the random pictures also have something in common
with the monotonous grid pictures : especially for large randomly generated
sets, the autocorrelation function is almost identical with the sets that consist
of monotonous grids, as the variation in the density is only noticeable on a
very small scale. For random sets larger than approximately 105 the difference
with monotonous grids of the same size is neglectable, as long as we are not
interested in events that take place on a very small scale and of course if
the average particle displacement during the simulation is not too large.
A special situation occurs for the random set when the mean particle displacement
lays between the smallest minimum interparticle distance ( SMI ) and the
average minimum interparticle ( AMI ) distance. In that case there will be a noticeable
difference in the number of collisions between random sets and monotonous sets
: for the monotonous sets we may say SMI = AMI so no collisions would be found, while
for the random sets SMI < AMI possibly resulting in a certain number of collisions.
Conclusion : On a large scale both monotonous grids and random sets
can be considered to be uniform as long as the displacement is small
enough.
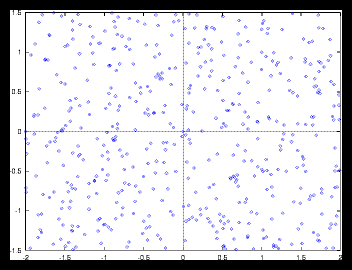
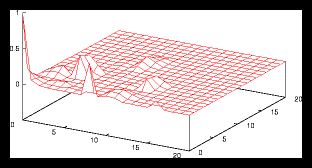
Figure 6: 500 particle random set with a few extra heavy masses causing non-uniformity.
Now let's see what happens with the autocorrelation if we have clearly non-uniform
distributions. These distributions can be created by adding some heavy masses
on a random set or a grid, by generating a few random sets at different locations
in space and merge them into one new set (clusters) or simply by using a set
with only a few particles.
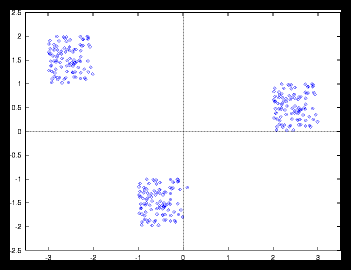
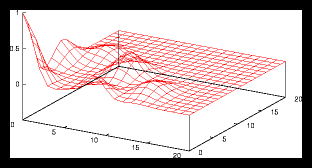
Figure 7: 300 particle set built by clustering 3 smaller random sets of 100 particles
each.
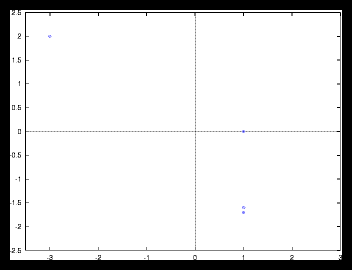
Figure 8: Autocorrelation for the 4 particle problem from figure 1.1.
Some examples are given in figures 6, 7 and 8. Note that in all these cases
the autocorrelation function drops very fast as soon we drift away from the
centre. This leads to the conclusion that the average value of the autocorrelation
function, especially around the centre, can provide us with the information
we are searching for.
I decided to use the average value of the autocorrelation function in both directions
from 0 to [1/4] of the grid dimension, to make the distinction clear enough. Far
away from the centre all pictures show that the autocorrelation is small, so
using these values only makes the distinction less clear. This property was
given the symbol T in the flowchart from figure 1.
In formula the rather simple definition I used for the discriminator T looks
like :
|
T = |
2
N
|
|
N/4
å
x = 1
|
A(x,0)+ |
2
N
|
|
N/4
å
y = 1
|
A(0,y) |
| (4) |
Where the autocorrelation function A(x,y) has to be rescaled to be always between
0 and 1.
Let's now summarise some values for T for true random and grid patterns and
compare them with the values found for the 3 non-uniform examples. First we'll
look at some gridpatterns generated by mkgrid (where SMI and AMI should
be essentially the same) :
|
Particles |
T |
SMI |
AMI
|
|
|
|
1024 |
0.512243 |
0.156250 |
0.156250
|
|
4096 |
0.505994 |
0.078125 |
0.078125
|
|
16384 |
0.500418 |
0.039062 |
0.039062
|
|
65536 |
0.489416 |
0.019531 |
0.019531
|
|
131072 |
0.496926 |
0.013810 |
0.013810 |
And now for some random sets generated by mkrnd (where SMI should be
of course less than AMI ) :
|
Particles |
T |
SMI |
AMI
|
|
|
|
1024 |
0.262296 |
0.022565 |
0.086043
|
|
4096 |
0.402716 |
0.004072 |
0.035512
|
|
16384 |
0.469110 |
0.003728 |
0.020702
|
|
65536 |
0.489416 |
0.000439 |
0.011138
|
|
131072 |
0.492564 |
0.001258 |
0.007941 |
It is clear from the table that the larger the number of particles, the closer
the value of T comes to the value found in the gridpattern examples (about T ~ 0.5 ).
In the first table we see a very slow drop in the value of T for large numbers
of particles. This is probably caused by the fact that I limited the dimension
of the mesh to a rather low maximum value to improve the performance.
Finally we also need the T values for the non-uniform examples, to see if the
discrimination made by using the value of T would be sufficient :
|
Particles |
T |
SMI |
AMI
|
|
|
|
4 |
0.007852 |
0.100000 |
1.568034
|
|
300 |
0.043666 |
0.002539 |
0.056207
|
|
500 |
0.003150 |
0.011130 |
0.076142 |
As shown in the table, the values of T are much smaller now. I decided to to
call every set with T > 0.4 a uniform distribution, every set with T < 0.1 a NON-uniform
distribution and all sets with 0.4 > T > 0.1 sets that need closer inspection. Of course
these values are just more or less arbitrary chosen, but nevertheless they are
good enough to serve the purpose: a rough discrimination between uniform
and non-uniform sets.
The sets belonging to the ``borderland'' could be done using Particle-Mesh as
long as the required accuracy is not too high, in the other case it would be
better to use Tree-Code, although this may take significantly longer as may
be seen in the results from the next chapter.
5. Implementation aspects
As usual there were several practical problems to be solved while building the
code. I will summon the most interesting ones hereafter :
- It should be possible to compare the factor T between all different types of
sets that should be analysed, no matter if they are large or small, uniform
or non-uniform. For that reason it was necessary to scale the autocorrelation
function back to alllow only values between 0 and 1 (vertical axis).
The same applies for the horizontal axis : we need the average value of the
summation, so it has to be scaled back with the number of particles N .
- The interparticle distance information ( SMI and AMI ) requires O(N2) operations. As
this would take too much time for the larger sets, I decided to use a randomiser
to draw a subset M of particles in this case and do the computation only for
this subset, reducing the number of computations to O(N·M) .
- A comparable problem as with the interparticle distance information occurs for
the computation of the initial acceleration to be able to determine the average
displacement. This was solved by using a Tree-Code like algorithm to calculate
this acceleration as soon as the number of particles exceeds a certain threshold.
- The average displacement contains important information about the system being
collisionless or not. But suppose the system as a whole would be translating
with a high speed. As this could make the information totally useless, I decided
to always transpose the problem to the Center-Of-Mass system. This of course
sets the total momentum to 0 in both directions.
- The meshspacing for the autocorrelation function is important : if it is too
large, the function may contain large fluctuations and if it is too small we
loose information (this is in fact exactly what happens with the large random
sets compared with the meshes !).
Also the overall meshsize is important: if the dimension is too small this may
cause aliasing, while if it is too large we loose information as the autocorrelation
drops very fast in that case.
Some examples of the analyser output are given in appendix .
6. Tunable parameters
00.00.0000
- [
-
-v]Verbose flag. This will cause extra information to be produced during the
analysis.
- [
-
-w < nr > ]Width of the mesh for the autocorrelation
function.Overrides the automatic computation of the grid and may result in non-comparable
values for T .
- [
-
-h < nr > ]Height of the mesh for the autocorrelation
function. See above.
- [
-
-n < nr > ]Number of mesh cells in one direction. See
above.
- [
-
-p]Write the autocorrelation function data into a file called ``autocorr.dat''
and plot the result on the screen using GNUplot.
- [
-
-l < nr > ]Threshold for large sets. Above this value
the analyser switches to use a random generator for the interparticle distance
information and a Tree-Code algorithm for the acceleration computation.
As many of the routines were taken from either the Particle-Mesh or the Tree-Code
programs, several of the changeable macros from these programs also apply here:
i.e. COLLISIONDISTANCE, LAMBDA etc..
Most of the other parameters that can be changed at compile time can be found
in the Initialize()routine:
- The Q factor for the Tree-Code part is currently set to 1 by default.
- The number of samples that is used to determine interparticle distance for large
sets is currently set to be R·logN , where the factor R has a default of 10.
- The threshold for the distinction between PP and PM/TC is set to 100 particles
by default.
Most of the other values set in the Initialize()routine are used to
give a reasonable advise after analysis. These numbers were mostly obtained
empirically.
7. Restrictions
Although this analyser heuristic seems to give reasonable clear indications
at this time, it isn't perfect and it may still contain many flaws, as it is
still very young and only little tested. Also many of the used characteristics
need further investigation. Nevertheless : it is much better than trying to
run a set through all 3 methods and see which one is best, if it is kept in
mind that the results only give rough indications.
As already said earlier in this chapter : especially the sets that are told
to be in the ``borderland'' need further examination before the final choice for
a certain method is made.
File translated from TEX by TTH, version 1.23.